In case you ever wondered what happens to scientific results after you share them with others, we wanted to give you a brief glimpse into what happened to the LilBUBome after we posted our data on bioRxiv two weeks ago. Continue reading
A manuscript with our LILBUBome results is online!
Dear All,
Today was a big day for us at the LilBUBome headquarters: after months of work, hundreds of emails back and forth and quite a few Skype meetings we made a preprint of our findings from LilBUB’s genome analysis public! If you are interested in reading it, you can find it here: 
Too much information – how to find something in BUBs genome
Hi everybody,
if you followed us here and here, you know that we have sequenced and mapped all of BUB’s genetic information.
Today we want to show you how we try to cut through all of the information that a genome holds to reveal the variants that we think made BUB BUB!
It’s all pretty exciting and also just a first glimpse on what we can find if we analyze even further! In any case. Have a nice weekend and fun watching – and as always: Let us know what you think or if you have any questions! We will answer. Pinky-promise (or in German: Großes Indianerehrenwort!)
Best
d
For the nerds of you: here’s a link to the software we used, it’s Platypus http://www.well.ox.ac.uk/platypus developed by Andy Rimmer and friends in Cambridge, UK.
For everyone – here’s the cool pic of the Hemingway Cats.
 source (http://www.hemingwayhome.com/cats/)
source (http://www.hemingwayhome.com/cats/)
Getting some info from BUB’s genome
Hi all,
we bring you a new episode from our videoblog. Today we show how we can retrieve info from BUB’s (or any other) genome.
We will use take raw data generated previously and use specific programs to visualize it.
And yes, the data shown in the video is BUB’s genetic code!!!!!!
Additional info:
Reading BUB’s DNA sequence: the raw data
Hi again,
Today we’re back with a new episode of the videolog, where I’ll explain what BUB’s DNA sequence looked like fresh off the sequencing machine. A couple of things to note: as our videos will get more and more technical please feel free to leave comments or to contact us if there’s anything you’d like us to clarify. Also, since we can’t include all the technical details, we’ll start leaving links to tools and papers together with the video, so you can follow up on stuff if you’re interested. Hope you enjoy!
More info about:
– the fastq format on wikipedia
– a technical note by Illumina about understanding quality scores
– a table for converting the characters in fastq files into an actual quality score
Starting the New Year with some LilBUBome updates
Hi All,
Happy New Year! We hope everyone has had a great start to 2016!
We know things have been a bit quiet around the LilBUBome, but the second half of last year was a busy time for us: Daniel, Dario and the sequencing team at the Max Planck Institute completed the sequencing of BUB’s DNA (we blogged about it here and here), and we’ve since been analyzing the data and following up on candidate mutations. We also did a bit of outreach, talking at various venues about sequencing BUB’s genome with the help of crowdfunding – and what an amazing experience has been to work with such a special cat and so many supporters!

In October we talked about the LilBUBome at the Sanger Institute postdoc retreat. While we were there we were allowed to have a quick look at their sequencing facility – and of course we took a picture for you!.
In addition, we’ve compiled a series of videos, which we’ll be releasing over the following weeks, where we try to explain what we did, why and how we did it and how you can go about sequencing a genome.
Today, let us introduce the first installment of the LilBUBome videolog. We hope you enjoy!
Sequencing the LilBUBome: choosing the “right” technology…
…and the technical parameters of our sequencing run.
Last week we gave you a brief and general overview of the different technologies that are available if you want to sequence a genome. So, how did we end up selecting Illumina sequencing? Since we’re frequently asked about our choice of technology, this post is dedicated to this topic. We’ll also discuss some of the technical details of our sequencing run, so if you’re interested in these kinds of things, keep reading.
Questions about the LilBUBome sequencing technology during our Reddit AMA.
Basically, our choice was an absolutely pragmatic one: We wanted to maximize our chances of finding something. Now, in terms of sequencing probably any of the available technologies would have given us data, but choosing the right one can make a difference when it comes to analyze certain parts of the genome. We decided to work with a method that was well-established in the institute where Daniel and Darío work. Their institute has an excellent sequencing facility, which routinely does Illumina sequencing and lots of complicated analysis, and so we have relied on their help and expertise throughout the hands-on part of the LilBUBome. Hence, why we chose Illumina.
As an added bonus there have been some unexpected benefits:
- As you may know, we are doing the LilBUBome as a labour of love, in addition to our regular work. We did not expect, however, that collaborating scientists would do the same. But they did! So we would like to use this opportunity to thank the staff of the Max Planck Institute’s Sequencing Facility, who have generously donated their hands and minds to the LilBUBome, completely free of charge! You already know Norbert from previous posts (for example here and here), but the same acknowledgment can be extended to the whole team. You are great!!!

Back row: Norbert Mages, Myriam Hochradel, Heiner Kuhl; Middle row: Daniela Roth, Sven Klages, Sonia Paturej, Bernd Timmermann; Front row: Hannes Cash, Ilona Hauenschild, Stefan Börno; Martin Kerick is missing in the pic, but we also love you 😉
- After we had completed crowdfunding and were preparing for the actual sequencing, Illumina kindly offered to provide us with free reagents for the sequencing. After some hesitation, we decided to accept the offer. Primarily, because it allowed us to upgrade the technology* – we were able to sequence Lil BUBs genome in more depth and quality, thus increasing our chances of success. In addition, we could use a method and a machine that’s much faster than the one we originally wanted to use (20 hours compared to 11 days!!!). And finally, this also means that we can make your donations go even further, to fully verify our findings from the sequencing (stay tuned!) with additional experiments.

—————————————
* And finally, here – as promised – the Technical details (aka TechTalk):
Sample preparation kit: Truseq sample prep for Next Seq Version2. Version 2 gives slightly longer reads and the coverage is much more even than with Version 1.
DNA fragmentation: We used a Covaris machine, according to the standard protocol provided by Illumina. We made 2 libraries (300bp and 500bp length) and then ligated adaptors. After adaptor ligation (Adaptors were roughly 120bp each), the libraries were run on a gel and the bands (sized 400-500bp for the 300bp library and 600-700bp for the 500bp library) were excised from the gel.
Seq machine: NextSeq 500. (Before the Illumina donation we would have used the HiSeq 2500. The NextSeq gave longer reads, and also the sequencing protocol was much faster (20 hours on the NextSeq vs 10-11 days of the Hiseq))
Read lenght : 2x 150bp. (If we would have used the HiSeq, we would have sequenced 2x 125bp reads. The longer reads from the NextSeq don’t improve coverage (40X), but they do allow for better assembly. Better assembly is crucial to detect small rearrangements.)
Paired end: Yes
The technology behind the LilBUBome: sequencing
Since we’ll be talking a lot about sequencing and sequencing results in the next few weeks, we’d like to start by telling you about the technology we used.
Generally speaking if you want to sequence something today there are two approaches: Sanger sequencing or next-generation sequencing (NGS) methods. NGS methods are the go-to methods if you want to sequence a complete genome (as we did with LilBUB), because the cost to sequence a single base (the building blocks of DNA) is much less and you can work at high-throughput (sequence a lot of DNA at the same time). Having said that it’s still a couple of thousand dollars per genome, so Sanger sequencing may be a better choice if you’re only interested in sequencing a small segment of DNA. Previously, we used Sanger sequencing to find the mutation that’s responsible for BUB’s polydactyly.
This time, to sequence the whole genome of Lil BUB we used NGS, though. More specifically, we used Illumina sequencing. Continue reading
Sequencing the LilBUBome – Video Diary part I
Hi All,
Despite the long silence, Daniel, Dario and I have been busy working on the LilBUBome, and we’ll be releasing more info in the next few weeks. As we reported last time, we had extracted high quality DNA from BUB’s blood sample. Now, we proceeded to the sequencing step!
Meanwhile, we’ve also been exploring new forms of communicating results with you, because we figured that reading long, technical blog posts might be pretty tedious. Here our first attempt at a LilBUBome video. Let us know what you think!!
A handful of BUB’s DNA
Some posts ago, we extracted some DNA from LilBUB to run a small test which revealed that BUB seems to be distantly related to Hemingway cats. However, to sequence her whole genome we require a bigger amount of DNA so we need to re-extract it from our blood sample (to remember the principles of DNA extraction check here).
But, how much DNA do you actually need to sequence the entire genome of an organism? Ideally 1 microgram should be enough. This is equivalent 0,000001 grams, which is the 50th part of a water drop. Pretty small amount, right? But in terms of our purpose more than enough!!!
After handling the sample to our sequencing facility, Norbert (one of the technicians working there) extracted BUB’s DNA. After the process he obtained a small volume of a water solution.

Norbert in action
But how do we know how much DNA is contained there and its purity? This is determined by the ability of the DNA to absorb light, which in scientific jargon we call Absorbance.
To measure these parameters, a small drop of our water solution is loaded on a spectrophotometer. This machine emits ultraviolet light (wavelength of 260 nm) that pass through our drop, and is received by a photodetector. The less light reaching the photodetector, the more DNA is included in our sample. This analysis revealed that our sample contains up to 4 micrograms of DNA (well done Norbert!!!).
The spectrophotometer is also capable of measuring other contaminants that could be present in our sample, derived from a not so clean extraction. These contaminants absorb different types of light (with different wavelengths). The Absorbance ratios between different types of light revealed no contamination of proteins (260/280=1,82) and organic solvents (260/230=2,1), confirming the high purity of our sample.
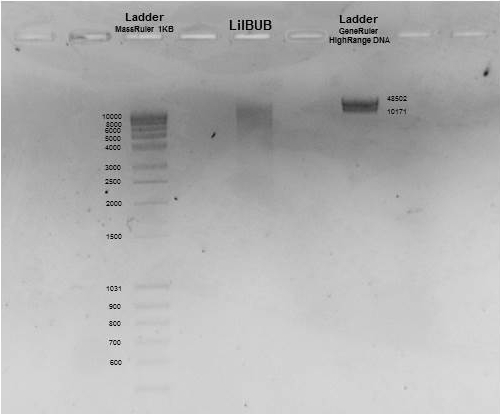
LilBUB´s DNA appears as a high molecular weight smeary band
Last, we checked the size of our DNA. For that purpose, a small part the sample is loaded in an agarose gel. When we apply voltage through the gel, our DNA will migrate from top to bottom. The bigger the size of our DNA, the slower it migrates. In our picture, you can appreciate that the size of our DNA is bigger that 10 kilobases, which means that it was minimally fragmented through the whole process. Taking into account that the genetic code is composed by bases (A, C, T or G) this means than, on average, we have long DNA stretches of more than 10000 bases!!!!
Next station, preparation of a DNA library for sequencing!!!!!
